
Abstract
The miR-17–92 cluster encoding 6 single mature miRNAs was identified a couple of years ago to contain the first oncogenic miRNAs. Now, one of these 6 miRNAs, miR-19 has been identified as the key responsible for this oncogenic activity. This in turn reduces PTEN levels and in consequence activates the AKT/mTOR pathway that is also prominently involved in modulation of organismal life spans. In contrast, miR-19 and other members of the miR-17–92 cluster are found to be commonly downregulated in several human replicative and organismal aging models. Taken together, these findings suggest that miR-19 and the other members of the miR-17–92 cluster might be important regulators on the cross-roads between aging and cancer. Therefore, we here briefly summarize how this cluster is transcriptionally regulated, which target mRNAs have been confirmed so far and how this might be linked to modulation of organismal life-spans.
Similar content being viewed by others


Aging, cancer and miR-17–92
It is not long ago that the first miRNA cluster has been identified with oncogenic potential and was therefore termed oncomiR-1 (He et al. 2005). Now, two recent reports have been able to pin down miR-19 as the key oncogenic miRNA of this cluster containing 6 miRNA members (Mu et al. 2009; Olive et al. 2009). Furthermore, the idea that miRNAs also play a role in aging is increasingly substantiated (Grillari and Grillari-Voglauer 2010; Bates et al. 2009). Recently, a large scale microRNA microarray analysis of 4 different cell types in replicative senescence and 3 different tissue types ex vivo representing organismal aging was performed (Hackl et al. 2010). Thereby, a common down-regulation of miR-17, 19b, 20a and miR-106a, members of the miR-17–92 and paralogous cluster, was found (see Table 1 for an overview of the clusters, their members, and their seed sequences). This indicates that this cluster represents one additional important player not only in the complex regulatory network of cell cycle and tumorigenesis, but also in aging, emphasising that these processes are intricately interwoven (Campisi 2003). Even more so, as miR-19 upregulation in cancer activates the AKT-mTOR pathway via PTEN silencing (Olive et al. 2009). It is tempting to speculate therefore, that decrease of miR-19 might lead to increased PTEN and in consequence repress AKT-mTOR, a pathway that has been clearly linked with modulation of life-span in a variety of model organisms (Kapahi et al. 2004; Blagosklonny 2007; Schieke and Finkel 2007) and even in mouse, where the mTOR inhibitor rapamycin leads to a life-span extension (Harrison et al. 2009).
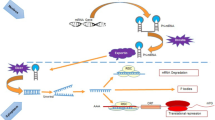
Thus, understanding the regulatory network of this cluster might well increase our knowledge on why advancing age is the largest single risk factor to develop cancer. Therefore, we here want to briefly summarize the current knowledge on the regulation loops of this cluster (Fig. 1) especially in regard to senescence that has been largely accepted as tumor suppressor mechanism in vivo (reviewed in Hornsby 2007; Sedivy 2007) and aging.

Overview on transcriptional regulation and target mRNAs of the miR-17–92 cluster
Transcriptional regulation of the miR-17–92 cluster
So far, c-MYC (O’Donnell et al. 2005), E2F1 and 3 (Petrocca et al. 2008b), as well as STAT3 (Brock et al. 2009) have been identified to transcriptionally activate the miR-17–92 cluster and paralogous clusters, while p53 represses it (Yan et al. 2009). However, MYC does not seem to change in senescence (Chang and Chen 1988; Seshadri and Campisi 1990), even though tumour cells enter senescence upon MYC inactivation (Wu et al. 2007). Similarly, STAT3 seems not to be involved, even if it might be expected to rise with senescence in response to the senescence-dependent increase of secreted IL-6 and IL-8 (Acosta et al. 2008) which are upstream activators of STAT3. Why in turn the miR-17–92 cluster is decreased instead of activated is unclear. STAT3 might be blocked at the post-translational level, since it is very susceptible to oxidation and is easily S-glutathionylated. In consequence of this modification, it is not activated by JAK anymore and does not translocate to the nucleus (Xie et al. 2009). High levels of S-glutathionylated, and thus inhibited STAT3 might be possible due to high levels of S-glutathione transferase P that are known to be present in senescent cells (Chang et al. 2005).
Thus, two responsible transcriptional regulators remain to most probably account for less miR-17–92 in senescence. Less E2F family members have been observed in senescent cells (Dimri et al. 1994), and p53, which is a decisive switch in aging and tumorigenesis (Rodier et al. 2007; Schmid et al. 2007) is increasingly active in senescence (Atadja et al. 1995; Kulju and Lehman 1995) and might thus contribute by actively repressing miR-17–92 (Yan et al. 2009).
Targets of the miR-17–92 cluster
Around 30 mRNA targets have been experimentally confirmed so far (Table 2), among them BCL2L11 (Bim), IRF, JNK2/MAPK9, MYCN, PKD1, PKD2, GAB1, RBL1, TSG101 (Cloonan et al. 2008), p63 (Manni et al. 2009), STAT3 and p38/Mapk14 (Carraro et al. 2009), the TGFβ signal pathway (Petrocca et al. 2008a), HIF-1α (Taguchi et al. 2008), or Rbl2/p130 (Wang et al. 2008), p57, p27 and p21 all involved in tumorigenesis and cell cycle control. Especially p21 transcription is well correlated with miR-17, 19b, 20a and miR-106a in the replicative and organismal aging model systems described above (Hackl et al. 2010).
Indeed, miR-17–92 suppression induces complete growth arrest in an anaplastic thyroid cancer cell model (Takakura et al. 2008). In contrast, overexpression of one of its members in mouse embryonic fibroblasts, miR-20a, induces senescence by reducing Leukemia/lymphoma Related Factor (LRF) levels (Poliseno et al. 2008), indicating that cell type specific responses are possible in response to miR-17–92. This is consistent with the notion that overexpression of miR-106a that derives from a paralogous cluster, targets p21 in human fibroblasts and trabecular meshwork cells (Li et al. 2009). Finally, overexpression of miR-17–92 inhibits generation of ROS and DNA damage in RB mutated tumor cells (Ebi et al. 2009).
It will be exciting to see if the opposite, reduction of miR-17–92 will result in more ROS and DNA damage, as well as block of tissue repair by inhibition of stem cell self renewal. All of these are well accepted driving forces of age-related functional decline.
Conclusion
It is still unclear, how and why miR-17–92 is downregulated during aging and senescence. Future work will have to reveal if it is cause or consequence and to what extent its downregulation functionally contributes to aging or even to tumor suppression during aging. In any case, members of this cluster might represent novel biomarkers of aging and the link between miR-17–92 and AKT/mTOR via PTEN might provide a novel regulatory loop of life span modulation.
References
Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, Takatsu Y, Melamed J, d’Adda di Fagagna F, Bernard D, Hernando E, Gil J (2008) Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133:1006–1018
Atadja P, Wong H, Garkavtsev I, Veillette C, Riabowol K (1995) Increased activity of p53 in senescing fibroblasts. Proc Natl Acad Sci USA 92:8348–8352
Bates DJ, Liang R, Li N, Wang E (2009) The impact of noncoding RNA on the biochemical and molecular mechanisms of aging. Biochim Biophys Acta 1790:970–979
Blagosklonny MV (2007) Paradoxes of aging. Cell Cycle 6:2997–3003
Brock M, Trenkmann M, Gay RE, Michel BA, Gay S, Fischler M, Ulrich S, Speich R, Huber LC (2009) Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circ Res 104:1184–1191
Campisi J (2003) Cancer and ageing: rival demons? Nat Rev Cancer 3:339–349
Carraro G, El-Hashash A, Guidolin D, Tiozzo C, Turcatel G, Young BM, De Langhe SP, Bellusci S, Shi W, Parnigotto PP, Warburton D (2009) miR-17 family of microRNAs controls FGF10-mediated embryonic lung epithelial branching morphogenesis through MAPK14 and STAT3 regulation of E-Cadherin distribution. Dev Biol 333:238–250
Chang ZF, Chen KY (1988) Regulation of ornithine decarboxylase and other cell cycle-dependent genes during senescence of IMR-90 human diploid fibroblasts. J Biol Chem 263:11431–11435
Chang MW, Grillari J, Mayrhofer C, Fortschegger K, Allmaier G, Marzban G, Katinger H, Voglauer R (2005) Comparison of early passage, senescent and hTERT immortalized endothelial cells. Exp Cell Res 309:121–136
Cloonan N, Brown MK, Steptoe AL, Wani S, Chan WL, Forrest AR, Kolle G, Gabrielli B, Grimmond SM (2008) The miR-17-5p microRNA is a key regulator of the G1/S phase cell cycle transition. Genome Biol 9:R127
Dimri GP, Hara E, Campisi J (1994) Regulation of two E2F-related genes in presenescent and senescent human fibroblasts. J Biol Chem 269:16180–16186
Ebi H, Sato T, Sugito N, Hosono Y, Yatabe Y, Matsuyama Y, Yamaguchi T, Osada H, Suzuki M, Takahashi T (2009) Counterbalance between RB inactivation and miR-17–92 overexpression in reactive oxygen species and DNA damage induction in lung cancers. Oncogene 28:3371–3379
Fontana L, Pelosi E, Greco P, Racanicchi S, Testa U, Liuzzi F, Croce CM, Brunetti E, Grignani F, Peschle C (2007) MicroRNAs 17-5p-20a-106a control monocytopoiesis through AML1 targeting and M-CSF receptor upregulation. Nat Cell Biol 9:775–787
Grillari J, Grillari-Voglauer R (2010) R Novel modulators of senescence, aging, and longevity: small non-coding RNAs enter the stage. Exp Gerontol 45:302–311
Hackl M, Brunner S, Fortschegger K, Schreiner C, Micutkova L, Mück C, Laschober GT, Lepperdinger G, Sampson N, Berger P, Herndler-Brandstetter D, Wieser M, Kühnel H, Strasser A, Breitenbach M, Rinnerthaler M, Eckhart L, Mildner M, Tschachler E, Papak C, Trost A, Bauer J, Scheideler M, Trajanoski Z, Grillari-Voglauer R, Grubeck-Loebenstein B, Jansen-Durr P, Grillari J (2010) miR-17, miR-19b, miR-20a and miR-106a are down-regulated in human aging. Aging Cell 9:291–296
Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA (2009) Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460:392–395
He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, Hammond SM (2005) A microRNA polycistron as a potential human oncogene. Nature 435:828–833
Hornsby PJ (2007) Senescence as an anticancer mechanism. J Clin Oncol 25:1852–1857
Hossain A, Kuo MT, Saunders GF (2006) Mir-17-5p regulates breast cancer cell proliferation by inhibiting translation of AIB1 mRNA. Mol Cell Biol 26:8191–8201
Ivanovska I, Ball AS, Diaz RL, Magnus JF, Kibukawa M, Schelter JM, Kobayashi SV, Lim L, Burchard J, Jackson AL, Linsley PS, Cleary MA (2008) MicroRNAs in the miR-106b family regulate p21/CDKN1A and promote cell cycle progression. Mol Cell Biol 28:2167–2174
Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S (2004) Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol 14:885–890
Kulju KS, Lehman JM (1995) Increased p53 protein associated with aging in human diploid fibroblasts. Exp Cell Res 217:336–345
Landais S, Landry S, Legault P, Rassart E (2007) Oncogenic potential of the miR-106-363 cluster and its implication in human T-cell leukemia. Cancer Res 67:5699–5707
Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB (2003) Prediction of mammalian microRNA targets. Cell 115:787–798
Li G, Luna C, Qiu J, Epstein DL, Gonzalez P (2009) Alterations in microRNA expression in stress-induced cellular senescence. Mech Ageing Dev 130:731–741
Manni I, Artuso S, Careccia S, Rizzo MG, Baserga R, Piaggio G, Sacchi A (2009) The microRNA miR-92 increases proliferation of myeloid cells and by targeting p63 modulates the abundance of its isoforms. Faseb J 23:3957–3960
Mu P, Han YC, Betel D, Yao E, Squatrito M, Ogrodowski P, de Stanchina E, D’Andrea A, Sander C, Ventura A (2009) Genetic dissection of the miR-17–92 cluster of microRNAs in Myc-induced B-cell lymphomas. Genes Dev 23:2806–2811
O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT (2005) c-Myc-regulated microRNAs modulate E2F1 expression. Nature 435:839–843
Ohgawara T, Kubota S, Kawaki H, Kondo S, Eguchi T, Kurio N, Aoyama E, Sasaki A, Takigawa M (2009) Regulation of chondrocytic phenotype by micro RNA 18a: involvement of Ccn2/Ctgf as a major target gene. FEBS Lett 583:1006–1010
Olive V, Bennett MJ, Walker JC, Ma C, Jiang I, Cordon-Cardo C, Li QJ, Lowe SW, Hannon GJ, He L (2009) miR-19 is a key oncogenic component of mir-17-92. Genes Dev 23:2839–2849
Patel N, Hoang D, Miller N, Ansaloni S, Huang Q, Rogers JT, Lee JC, Saunders AJ (2008) MicroRNAs can regulate human APP levels. Mol Neurodegener 3:10
Petrocca F, Vecchione A, Croce CM (2008a) Emerging role of miR-106b-25/miR-17-92 clusters in the control of transforming growth factor beta signaling. Cancer Res 68:8191–8194
Petrocca F, Visone R, Onelli MR, Shah MH, Nicoloso MS, de Martino I, Iliopoulos D, Pilozzi E, Liu CG, Negrini M, Cavazzini L, Volinia S, Alder H, Ruco LP, Baldassarre G, Croce CM, Vecchione A (2008b) E2F1-regulated microRNAs impair TGFbeta-dependent cell-cycle arrest and apoptosis in gastric cancer. Cancer Cell 13:272–286
Pichiorri F, Suh SS, Ladetto M, Kuehl M, Palumbo T, Drandi D, Taccioli C, Zanesi N, Alder H, Hagan JP, Munker R, Volinia S, Boccadoro M, Garzon R, Palumbo A, Aqeilan RI, Croce CM (2008) MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis. Proc Natl Acad Sci USA 105:12885–12890
Pickering MT, Stadler BM, Kowalik TF (2009) miR-17 and miR-20a temper an E2F1-induced G1 checkpoint to regulate cell cycle progression. Oncogene 28:140–145
Poliseno L, Pitto L, Simili M, Mariani L, Riccardi L, Ciucci A, Rizzo M, Evangelista M, Mercatanti A, Pandolfi PP, Rainaldi G (2008) The proto-oncogene LRF is under post-transcriptional control of MiR-20a: implications for senescence. PLoS One 3:e2542
Rodier F, Campisi J, Bhaumik D (2007) Two faces of p53: aging and tumor suppression. Nucleic Acids Res 35:7475–7484
Sampath D, Calin GA, Puduvalli VK, Gopisetty G, Taccioli C, Liu CG, Ewald B, Liu C, Keating MJ, Plunkett W (2009) Specific activation of microRNA106b enables the p73 apoptotic response in chronic lymphocytic leukemia by targeting the ubiquitin ligase Itch for degradation. Blood 113:3744–3753
Schieke SM, Finkel T (2007) TOR and aging: less is more. Cell Metab 5:233–235
Schmid G, Kramer MP, Maurer M, Wandl S, Wesierska-Gadek J (2007) Cellular and organismal ageing: role of the p53 tumor suppressor protein in the induction of transient and terminal senescence. J Cell Biochem 101:1355–1369
Sedivy JM (2007) Telomeres limit cancer growth by inducing senescence: long-sought in vivo evidence obtained. Cancer Cell 11:389–391
Sengupta S, Nie J, Wagner RJ, Yang C, Stewart R, Thomson JA (2009) MicroRNA 92b controls the G1/S checkpoint gene p57 in human embryonic stem cells. Stem Cells 27:1524–1528
Seshadri T, Campisi J (1990) Repression of c-fos transcription and an altered genetic program in senescent human fibroblasts. Science 247:205–209
Taguchi A, Yanagisawa K, Tanaka M, Cao K, Matsuyama Y, Goto H, Takahashi T (2008) Identification of hypoxia-inducible factor-1 alpha as a novel target for miR-17-92 microRNA cluster. Cancer Res 68:5540–5545
Takakura S, Mitsutake N, Nakashima M, Namba H, Saenko VA, Rogounovitch TI, Nakazawa Y, Hayashi T, Ohtsuru A, Yamashita S (2008) Oncogenic role of miR-17-92 cluster in anaplastic thyroid cancer cells. Cancer Sci 99:1147–1154
Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, Prueitt RL, Yanaihara N, Lanza G, Scarpa A, Vecchione A, Negrini M, Harris CC, Croce CM (2006) A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci USA 103:2257–2261
Wang Q, Li YC, Wang J, Kong J, Qi Y, Quigg RJ, Li X (2008) miR-17-92 cluster accelerates adipocyte differentiation by negatively regulating tumor-suppressor Rb2/p130. Proc Natl Acad Sci USA 105:2889–2894
Wu CH, van Riggelen J, Yetil A, Fan AC, Bachireddy P, Felsher DW (2007) Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc Natl Acad Sci USA 104:13028–13033
Xie Y, Kole S, Precht P, Pazin MJ, Bernier M (2009) S-glutathionylation impairs signal transducer and activator of transcription 3 activation and signaling. Endocrinology 150:1122–1131
Yan HL, Xue G, Mei Q, Wang YZ, Ding FX, Liu MF, Lu MH, Tang Y, Yu HY, Sun SH (2009) Repression of the miR-17–92 cluster by p53 has an important function in hypoxia-induced apoptosis. Embo J 28:2719–2732
Ye W, Lv Q, Wong CK, Hu S, Fu C, Hua Z, Cai G, Li G, Yang BB, Zhang Y (2008) The effect of central loops in miRNA:MRE duplexes on the efficiency of miRNA-mediated gene regulation. PLoS One 3:e1719
Yu Z, Wang C, Wang M, Li Z, Casimiro MC, Liu M, Wu K, Whittle J, Ju X, Hyslop T, McCue P, Pestell RG (2008) A cyclin D1/microRNA 17/20 regulatory feedback loop in control of breast cancer cell proliferation. J Cell Biol 182:509–517
Acknowledgements
Work in the Grillari lab is supported by GEN-AU Project 820982 “Non-coding RNAs”, FWF S93-06, by CE.R.I.E.S., and the Herzfelder’sche Familienstiftung.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Grillari, J., Hackl, M. & Grillari-Voglauer, R. miR-17–92 cluster: ups and downs in cancer and aging. Biogerontology 11, 501–506 (2010). https://doi.org/10.1007/s10522-010-9272-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10522-010-9272-9