Mitochondrial Signatures in Circulating Extracellular Vesicles of Older Adults with Parkinson’s Disease: Results from the EXosomes in PArkiNson’s Disease (EXPAND) Study

 , , ,
, , ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Participants
2.2. Blood Sampling and Serum Separation
2.3. Isolation and Characterization of Small Extracellular Vesicles/Exosomes
2.3.1. Purification of Small Extracellular Vesicles/Exosomes
2.3.2. Western Immunoblot Analysis of Small Extracellular Vesicles
2.4. Measurement of Serum Concentrations of Inflammatory and Neurotrophic Biomolecules
2.5. Statistical Analysis
2.5.1. Partial Least Squares Discriminant Analysis
2.5.2. Soft Independent Modeling of Class Analogies
3. Results
3.1. Characteristics of the Study Participants
3.2. Characterization of Small Extracellular Vesicles in Serum of Participants with and without Parkinson’s Disease
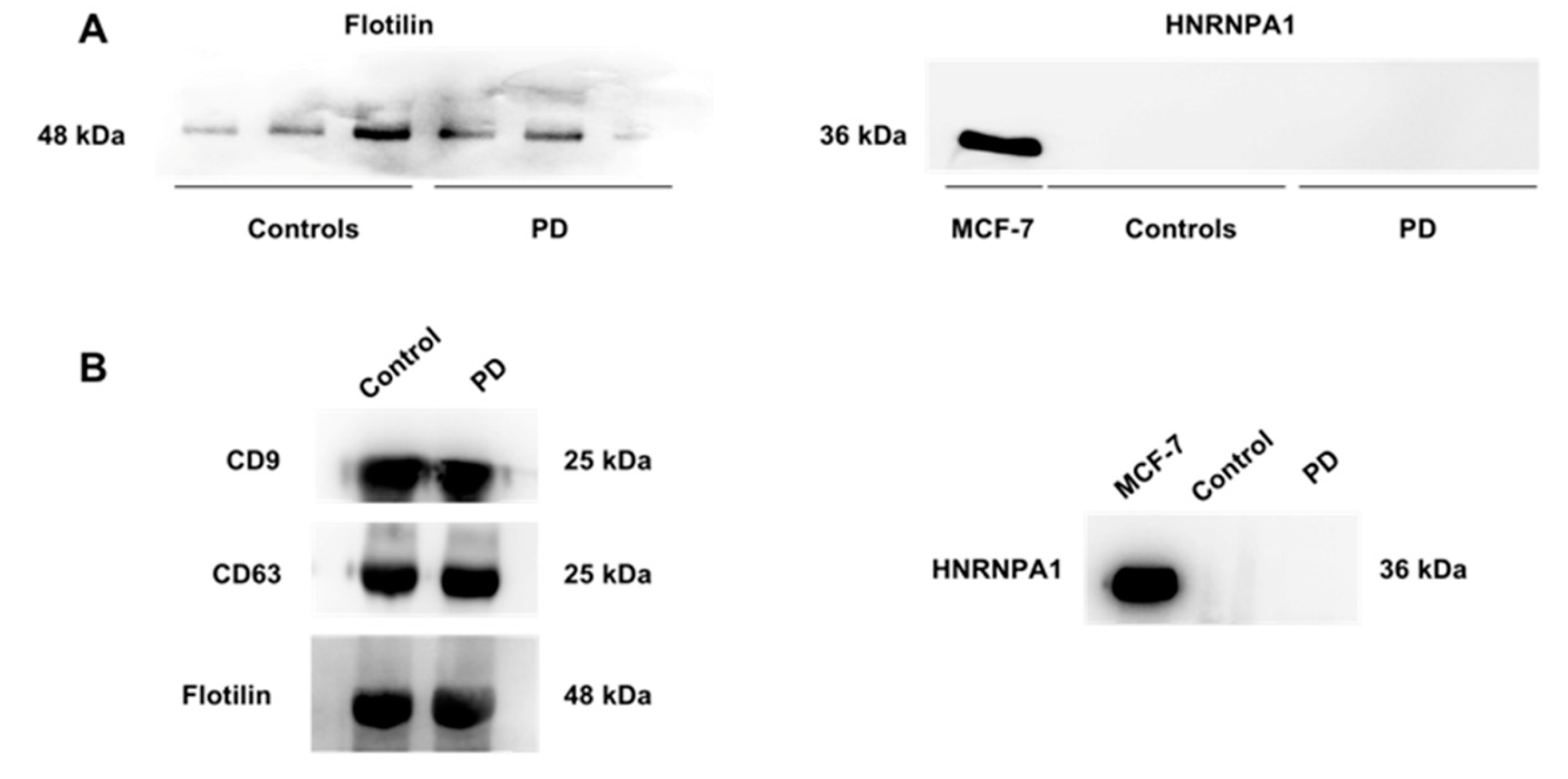
3.2.1. Characterization of Small Extracellular Vesicles
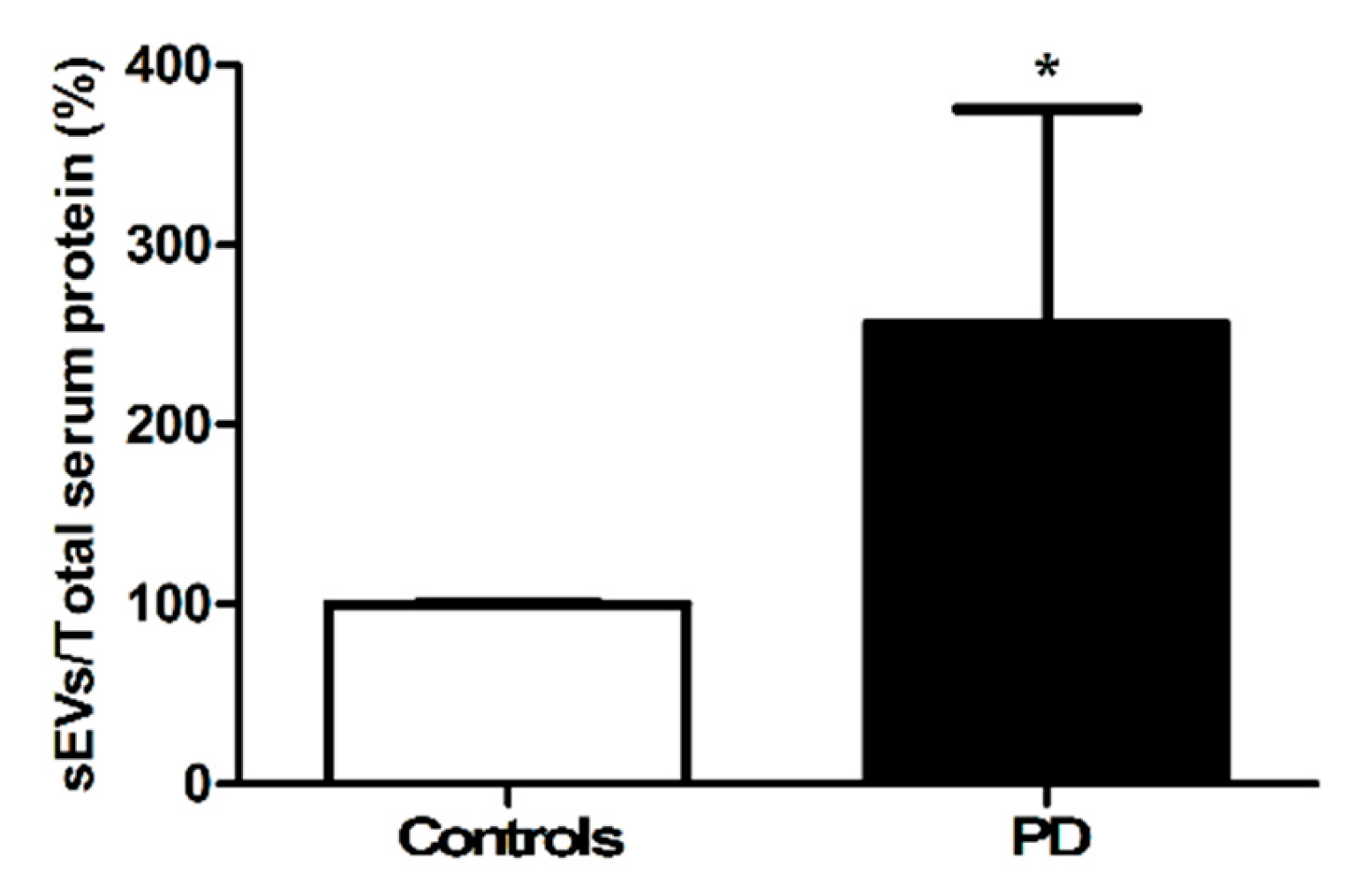
3.2.2. Quantification of the Amount of Circulating Small Extracellular Vesicles
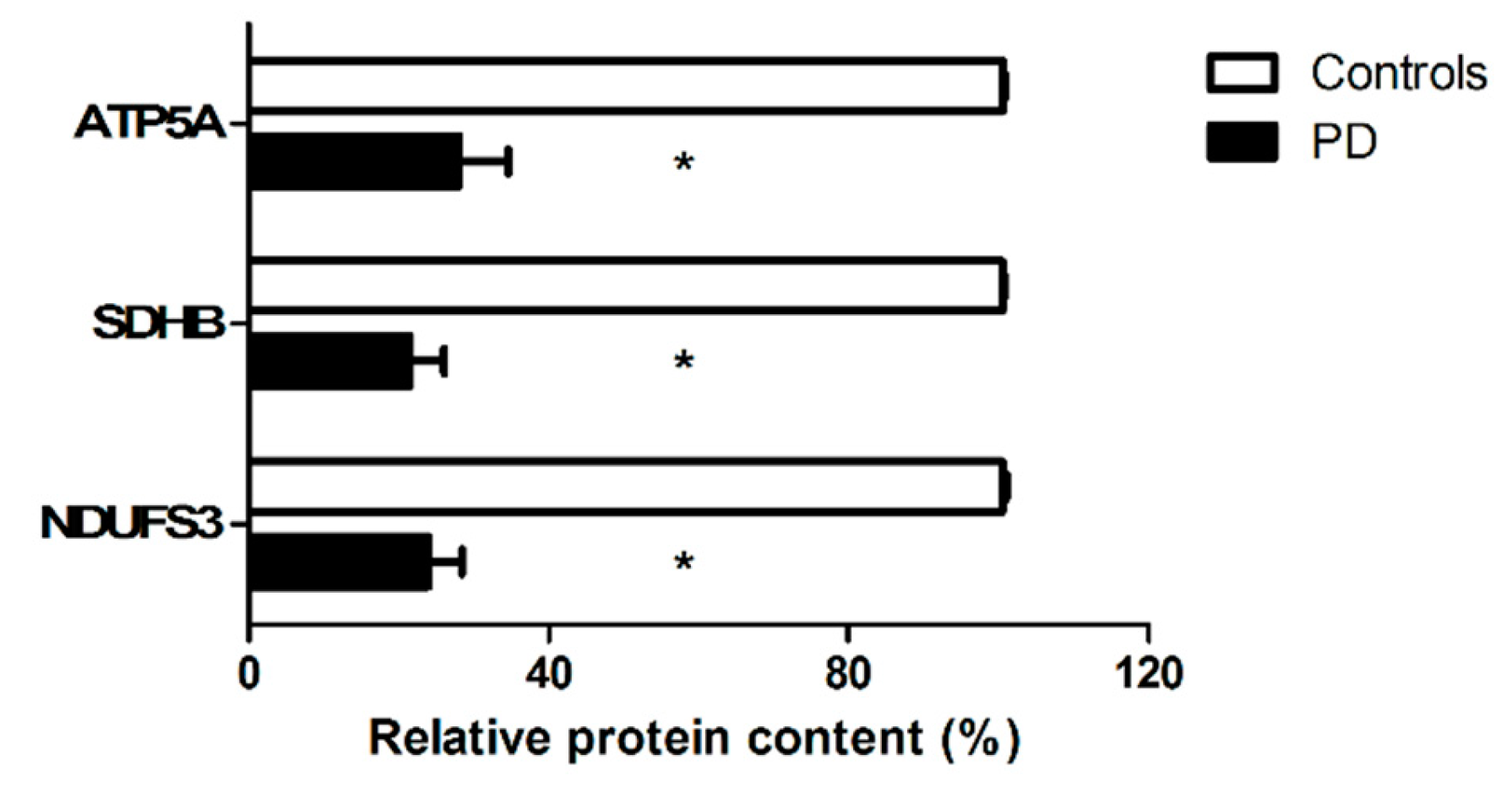
3.2.3. Characterization of the Cargo of Small Extracellular Vesicles
3.3. Identification of a Biomolecular Signature of Parkinson’s Disease by Partial Least Squares Discriminant Analysis
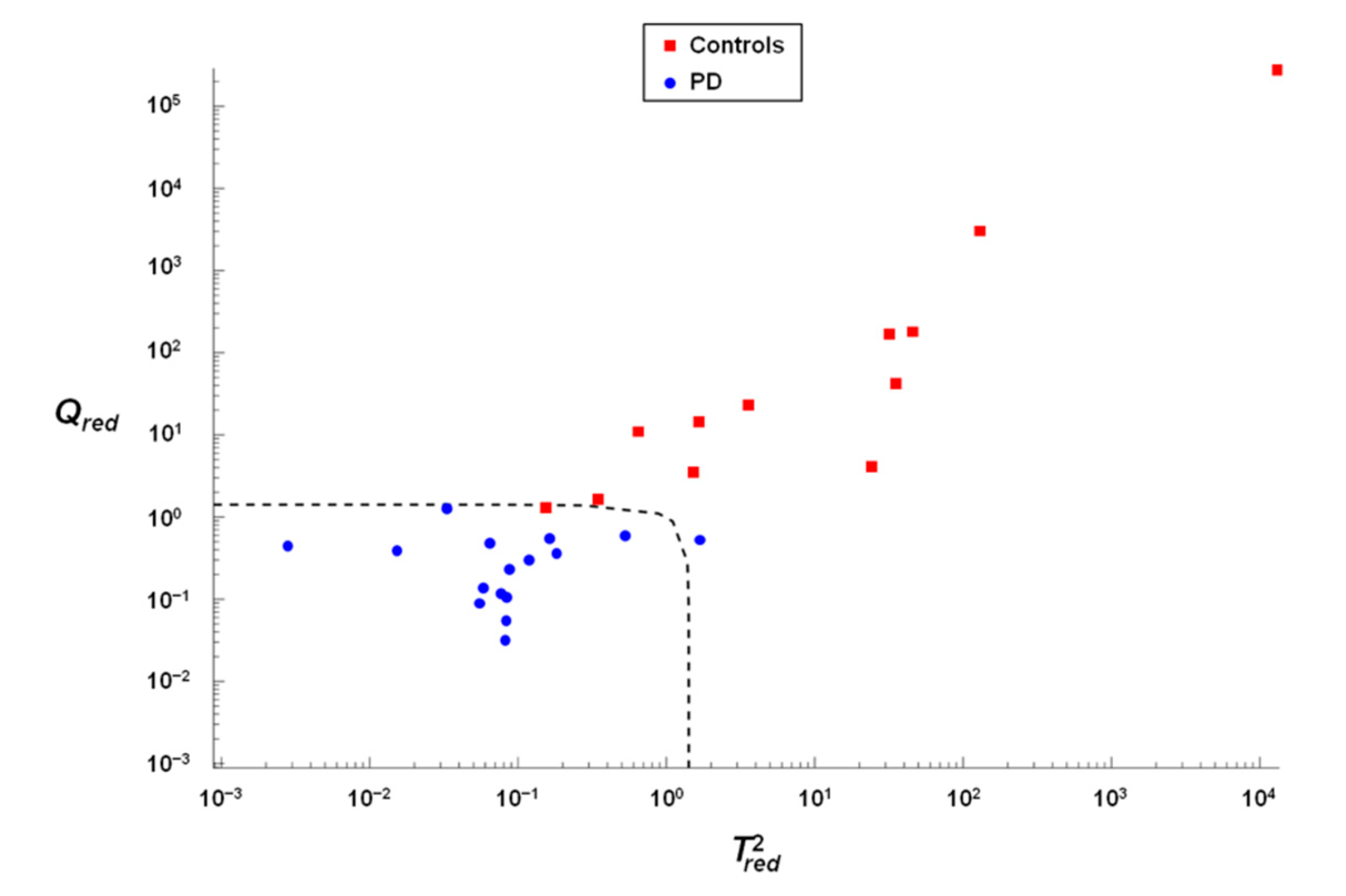
3.4. Verification of the Accuracy of Classification by Soft Independent Modeling of Class Analogies
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Brooks, D.J. The early diagnosis of Parkinson’s disease. Ann. Neurol. 1998, 44, S10–S18. [Google Scholar] [CrossRef] [PubMed]
- GBD 2016 Parkinson’s Disease Collaborators. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Alexander, G.E. Biology of Parkinson’s disease: Pathogenesis and pathophysiology of a multisystem neurodegenerative disorder. Dialogues Clin. Neurosci. 2004, 6, 259–280. [Google Scholar]
- Maiti, P.; Manna, J.; Dunbar, G.L. Current understanding of the molecular mechanisms in Parkinson’s disease: Targets for potential treatments. Transl. Neurodegener. 2017, 6, 28. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-S.; Davis, R.L.; Sue, C.M. Mitochondrial dysfunction in Parkinson’s disease: New mechanistic insights and therapeutic perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 21. [Google Scholar] [CrossRef] [Green Version]
- Melo, T.Q.; Copray, S.J.C.V.M.; Ferrari, M.F.R. Alpha-synuclein toxicity on protein quality control, mitochondria and endoplasmic reticulum. Neurochem. Res. 2018, 43, 2212–2223. [Google Scholar] [CrossRef]
- Cho, B.; Kim, T.; Huh, Y.-J.; Lee, J.; Lee, Y.-I. Amelioration of mitochondrial quality control and proteostasis by natural compounds in parkinson’s disease models. Int. J. Mol. Sci. 2019, 20, 5208. [Google Scholar] [CrossRef] [Green Version]
- White, A.J.; Wijeyekoon, R.S.; Scott, K.M.; Gunawardana, N.P.; Hayat, S.; Solim, I.H.; McMahon, H.T.; Barker, R.A.; Williams-Gray, C.H. The peripheral inflammatory response to alpha-synuclein and endotoxin in parkinson’s disease. Front. Neurol. 2018, 9, 946. [Google Scholar] [CrossRef]
- Picca, A.; Calvani, R.; Landi, G.; Marini, F.; Biancolillo, A.; Gervasoni, J.; Persichilli, S.; Primiano, A.; Urbani, A.; Bossola, M.; et al. Circulating amino acid signature in older people with Parkinson’s disease: A metabolic complement to the EXosomes in PArkiNson Disease (EXPAND) study. Exp. Gerontol. 2019, 128, 110766. [Google Scholar] [CrossRef]
- Picca, A.; Lezza, A.M.S.; Leeuwenburgh, C.; Pesce, V.; Calvani, R.; Landi, F.; Bernabei, R.; Marzetti, E. Fueling inflamm-aging through mitochondrial dysfunction: Mechanisms and molecular targets. Int. J. Mol. Sci. 2017, 18, 933. [Google Scholar] [CrossRef] [PubMed]
- Franco-Iborra, S.; Vila, M.; Perier, C. Mitochondrial quality control in neurodegenerative diseases: Focus on Parkinson’s disease and Huntington’s disease. Front. Neurosci. 2018, 12, 342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meldolesi, J. Exosomes and ectosomes in intercellular communication. Curr. Biol. 2018, 28, R435–R444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Cocucci, E.; Meldolesi, J. Ectosomes and exosomes: Shedding the confusion between extracellular vesicles. Trends Cell Biol. 2015, 25, 364–372. [Google Scholar] [CrossRef]
- Casella, G.; Colombo, F.; Finardi, A.; Descamps, H.; Ill-Raga, G.; Spinelli, A.; Podini, P.; Bastoni, M.; Martino, G.; Muzio, L.; et al. Extracellular vesicles containing IL-4 modulate neuroinflammation in a mouse model of multiple sclerosis. Mol. Ther. 2018, 26, 2107–2118. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Guerra, F.; Calvani, R.; Bucci, C.; Lo Monaco, M.R.; Bentivoglio, A.R.; Coelho-Júnior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial dysfunction and aging: Insights from the analysis of extracellular vesicles. Int. J. Mol. Sci. 2019, 20, 805. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Lezza, A.M.S.; Leeuwenburgh, C.; Pesce, V.; Calvani, R.; Bossola, M.; Manes-Gravina, E.; Landi, F.; Bernabei, R.; Marzetti, E. Circulating mitochondrial DNA at the crossroads of mitochondrial dysfunction and inflammation during aging and muscle wasting disorders. Rejuvenat. Res. 2018, 21, 350–359. [Google Scholar] [CrossRef]
- Schifferli, J.A. Microvesicles are messengers. Semin. Immunopathol. 2011, 33, 393–394. [Google Scholar] [CrossRef]
- Yoon, Y.J.; Kim, O.Y.; Gho, Y.S. Extracellular vesicles as emerging intercellular communicasomes. BMB Rep. 2014, 47, 531–539. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Bucci, C.; Lo Monaco, M.R.; Bentivoglio, A.R.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial-derived vesicles as candidate biomarkers in Parkinson’s disease: Rationale, design and methods of the EXosomes in PArkiNson Disease (EXPAND) study. Int. J. Mol. Sci. 2019, 20, 2373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, A.J.; Daniel, S.E.; Kilford, L.; Lees, A.J. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: A clinico-pathological study of 100 cases. J. Neurol. Neurosurg. Psychiatry 1992, 55, 181–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 30, 3.22.1–3.22.29. [Google Scholar] [CrossRef] [PubMed]
- Guerra, F.; Paiano, A.; Migoni, D.; Girolimetti, G.; Perrone, A.M.; De Iaco, P.; Fanizzi, F.P.; Gasparre, G.; Bucci, C. Modulation of RAB7A protein expression determines resistance to cisplatin through late endocytic pathway impairment and extracellular vesicular secretion. Cancers 2019, 11, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Théry, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef] [Green Version]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Marzetti, E.; Picca, A.; Marini, F.; Biancolillo, A.; Coelho-Junior, H.J.; Gervasoni, J.; Bossola, M.; Cesari, M.; Onder, G.; Landi, F.; et al. Inflammatory signatures in older persons with physical frailty and sarcopenia: The frailty “cytokinome” at its core. Exp. Gerontol. 2019, 122, 129–138. [Google Scholar] [CrossRef]
- Picca, A.; Ponziani, F.R.; Calvani, R.; Marini, F.; Biancolillo, A.; Coelho-Junior, H.J.; Gervasoni, J.; Primiano, A.; Putignani, L.; Del Chierico, F.; et al. Gut Microbial, Inflammatory and Metabolic Signatures in Older People with Physical Frailty and Sarcopenia: Results from the BIOSPHERE Study. Nutrients 2019, 12, 65. [Google Scholar] [CrossRef] [Green Version]
- Ståhle, L.; Wold, S. Partial least squares analysis with cross-validation for the two-class problem: A Monte Carlo study. J. Chemom. 1987, 1, 185–196. [Google Scholar] [CrossRef]
- Westerhuis, J.A.; Hoefsloot, H.C.J.; Smit, S.; Vis, D.J.; Smilde, A.K.; van Velzen, E.J.J.; van Duijnhoven, J.P.M.; van Dorsten, F.A. Assessment of PLSDA cross validation. Metabolomics 2008, 4, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Smit, S.; van Breemen, M.J.; Hoefsloot, H.C.J.; Smilde, A.K.; Aerts, J.M.F.G.; de Koster, C.G. Assessing the statistical validity of proteomics based biomarkers. Anal. Chim. Acta 2007, 592, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Marzetti, E.; Landi, F.; Marini, F.; Cesari, M.; Buford, T.W.; Manini, T.M.; Onder, G.; Pahor, M.; Bernabei, R.; Leeuwenburgh, C.; et al. Patterns of circulating inflammatory biomarkers in older persons with varying levels of physical performance: A partial least squares-discriminant analysis approach. Front. Med. 2014, 1, 27. [Google Scholar] [CrossRef] [PubMed]
- Wold, S.; Martens, H.; Wold, H. The multivariate calibration problem in chemistry solved by the PLS method. In Lecture Notes in Mathematics; Kågström, B., Ruhe, A., Eds.; Springer: Berlin, Germany, 1983; Volume 973, pp. 286–293. [Google Scholar]
- Calvani, R.; Picca, A.; Marini, F.; Biancolillo, A.; Gervasoni, J.; Persichilli, S.; Primiano, A.; Coelho-Junior, H.J.; Bossola, M.; Urbani, A.; et al. A Distinct Pattern of Circulating Amino Acids Characterizes Older Persons with Physical Frailty and Sarcopenia: Results from the BIOSPHERE Study. Nutrients 2018, 10, 1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wold, S. Pattern recognition by means of disjoint principal components models. Pattern Recognit. 1976, 8, 127–139. [Google Scholar] [CrossRef]
- Wold, S.; Sjöström, M. SIMCA: A method for analyzing chemical data in terms of similarity and analogy. Chemom. Theory Appl. 1977, 243–282. [Google Scholar] [CrossRef]
- Larsen, S.B.; Hanss, Z.; Krüger, R. The genetic architecture of mitochondrial dysfunction in Parkinson’s disease. Cell Tissue Res. 2018, 373, 21–37. [Google Scholar] [CrossRef] [Green Version]
- Guerra, F.; Girolimetti, G.; Beli, R.; Mitruccio, M.; Pacelli, C.; Ferretta, A.; Gasparre, G.; Cocco, T.; Bucci, C. Synergistic effect of mitochondrial and lysosomal dysfunction in Parkinson’s disease. Cells 2019, 8, 452. [Google Scholar] [CrossRef] [Green Version]
- Deleidi, M.; Gasser, T. The role of inflammation in sporadic and familial Parkinson’s disease. Cell. Mol. Life Sci. 2013, 70, 4259–4273. [Google Scholar] [CrossRef]
- Collins, L.M.; Toulouse, A.; Connor, T.J.; Nolan, Y.M. Contributions of central and systemic inflammation to the pathophysiology of Parkinson’s disease. Neuropharmacology 2012, 62, 2154–2168. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, A.; McLelland, G.-L.; Fon, E.A.; McBride, H.M. A new pathway for mitochondrial quality control: Mitochondrial-derived vesicles. EMBO J. 2014, 33, 2142–2156. [Google Scholar] [CrossRef] [Green Version]
- Verstraeten, A.; Theuns, J.; Van Broeckhoven, C. Progress in unraveling the genetic etiology of Parkinson disease in a genomic era. Trends Genet. 2015, 31, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Kirby, D.M.; Salemi, R.; Sugiana, C.; Ohtake, A.; Parry, L.; Bell, K.M.; Kirk, E.P.; Boneh, A.; Taylor, R.W.; Dahl, H.H.M.; et al. NDUFS6 mutations are a novel cause of lethal neonatal mitochondrial complex I deficiency. J. Clin. Investig. 2004, 114, 837–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFarland, R.; Kirby, D.M.; Fowler, K.J.; Ohtake, A.; Ryan, M.T.; Amor, D.J.; Fletcher, J.M.; Dixon, J.W.; Collins, F.A.; Turnbull, D.M.; et al. De novo mutations in the mitochondrial ND3 gene as a cause of infantile mitochondrial encephalopathy and complex I deficiency. Ann. Neurol. 2004, 55, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.D.; Parks, J.K.; Swerdlow, R.H. Complex I deficiency in Parkinson’s disease frontal cortex. Brain Res. 2008, 1189, 215–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amo, T.; Saiki, S.; Sawayama, T.; Sato, S.; Hattori, N. Detailed analysis of mitochondrial respiratory chain defects caused by loss of PINK1. Neurosci. Lett. 2014, 580, 37–40. [Google Scholar] [CrossRef]
- Bouvier-Müller, A.; Ducongé, F. Nucleic acid aptamers for neurodegenerative diseases. Biochimie 2018, 145, 73–83. [Google Scholar] [CrossRef]
- Kharitonenkov, A.; DiMarchi, R. Fibroblast growth factor 21 night watch: Advances and uncertainties in the field. J. Intern. Med. 2017, 281, 233–246. [Google Scholar] [CrossRef]
- Restelli, L.M.; Oettinghaus, B.; Halliday, M.; Agca, C.; Licci, M.; Sironi, L.; Savoia, C.; Hench, J.; Tolnay, M.; Neutzner, A.; et al. Neuronal Mitochondrial Dysfunction activates the integrated stress response to induce fibroblast growth factor 21. Cell Rep. 2018, 24, 1407–1414. [Google Scholar] [CrossRef] [Green Version]
- Kustrimovic, N.; Comi, C.; Magistrelli, L.; Rasini, E.; Legnaro, M.; Bombelli, R.; Aleksic, I.; Blandini, F.; Minafra, B.; Riboldazzi, G.; et al. Parkinson’s disease patients have a complex phenotypic and functional Th1 bias: Cross-sectional studies of CD4+ Th1/Th2/T17 and Treg in drug-naïve and drug-treated patients. J. Neuroinflammat. 2018, 15, 205. [Google Scholar] [CrossRef]
- Elyaman, W.; Khoury, S.J. Th9 cells in the pathogenesis of EAE and multiple sclerosis. Semin. Immunopathol. 2017, 39, 79–87. [Google Scholar] [CrossRef]
- Elyaman, W.; Bradshaw, E.M.; Uyttenhove, C.; Dardalhon, V.; Awasthi, A.; Imitola, J.; Bettelli, E.; Oukka, M.; van Snick, J.; Renauld, J.-C.; et al. IL-9 induces differentiation of TH17 cells and enhances function of FoxP3+ natural regulatory T cells. Proc. Natl. Acad. Sci. USA 2009, 106, 12885–12890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schröder, J.B.; Pawlowski, M.; Meyer Zu Hörste, G.; Gross, C.C.; Wiendl, H.; Meuth, S.G.; Ruck, T.; Warnecke, T. Immune cell activation in the cerebrospinal fluid of patients with Parkinson’s disease. Front. Neurol. 2018, 9, 1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, H.Y.; Cesari, M.; Anton, S.; Marzetti, E.; Giovannini, S.; Seo, A.Y.; Carter, C.; Yu, B.P.; Leeuwenburgh, C. Molecular inflammation: Underpinnings of aging and age-related diseases. Ageing Res. Rev. 2009, 8, 18–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Justice, J.N.; Ferrucci, L.; Newman, A.B.; Aroda, V.R.; Bahnson, J.L.; Divers, J.; Espeland, M.A.; Marcovina, S.; Pollak, M.N.; Kritchevsky, S.B.; et al. A framework for selection of blood-based biomarkers for geroscience-guided clinical trials: Report from the TAME Biomarkers Workgroup. GeroScience 2018, 40, 419–436. [Google Scholar] [CrossRef]
- Qiu, X.; Xiao, Y.; Wu, J.; Gan, L.; Huang, Y.; Wang, J. C-reactive orotein and risk of Parkinson’s disease: A systematic review and meta-analysis. Front. Neurol. 2019, 10, 384. [Google Scholar] [CrossRef]
- Perrin, F.E.; Lacroix, S.; Avilés-Trigueros, M.; David, S. Involvement of monocyte chemoattractant protein-1, macrophage inflammatory protein-1alpha and interleukin-1beta in Wallerian degeneration. Brain 2005, 128, 854–866. [Google Scholar] [CrossRef] [Green Version]
- Ahmadi Rastegar, D.; Ho, N.; Halliday, G.M.; Dzamko, N. Parkinson’s progression prediction using machine learning and serum cytokines. NPJ Park. Dis. 2019, 5, 14. [Google Scholar] [CrossRef]
- Probert, L. TNF and its receptors in the CNS: The essential, the desirable and the deleterious effects. Neuroscience 2015, 302, 2–22. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, S.L.; Bowers, W.J. Tumor necrosis factor-alpha and the roles it plays in homeostatic and degenerative processes within the central nervous system. J. Neuroimmune Pharmacol. 2012, 7, 42–59. [Google Scholar] [CrossRef]
- Ferger, B.; Leng, A.; Mura, A.; Hengerer, B.; Feldon, J. Genetic ablation of tumor necrosis factor-alpha (TNF-alpha) and pharmacological inhibition of TNF-synthesis attenuates MPTP toxicity in mouse striatum. J. Neurochem. 2004, 89, 822–833. [Google Scholar] [CrossRef]
- Nishimura, M.; Mizuta, I.; Mizuta, E.; Yamasaki, S.; Ohta, M.; Kaji, R.; Kuno, S. Tumor necrosis factor gene polymorphisms in patients with sporadic Parkinson’s disease. Neurosci. Lett. 2001, 311, 1–4. [Google Scholar] [CrossRef]
- Peter, I.; Dubinsky, M.; Bressman, S.; Park, A.; Lu, C.; Chen, N.; Wang, A. Anti-tumor necrosis factor therapy and incidence of Parkinson disease among patients with inflammatory bowel disease. JAMA Neurol. 2018, 75, 939–946. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Manufacturer and Catalog Number | Type | Species | Dilution | Detected Band MW (kDa) |
|---|---|---|---|---|---|
| ATP5A (complex V) MTCOI (complex IV) NDUFB8 (complex I) SDHB (complex II) UQCRC2 (complex III) | Abcam (Cambridge, MA, USA) ab1104413 | Monoclonal | Mouse | 1:250 | 55 40 20 30 48 |
| CD9 | Santa Cruz Biotechnology (Santa Cruz, CA, USA) (sc-13118) | Monoclonal | Mouse | 1:200 | 25 |
| CD63 | Santa Cruz Biotechnology (sc-5275) | Monoclonal | Mouse | 1:200 | 26 |
| CD81 | Santa Cruz Biotechnology (sc-166020) | Monoclonal | Mouse | 1:200 | 25 |
| NDUFS3 (complex I) | Santa Cruz Biotechnology (sc-374283) | Monoclonal | Mouse | 1:200 | 25 |
| Flotilin | Santa Cruz Biotechnology (sc-74566) | Monoclonal | Mouse | 1:200 | 48 |
| HNRNPA1 | Santa Cruz Biotechnology (sc-32301) | Monoclonal | Mouse | 1:1000 | 36 |
| Biomarker Class | Assayed Biomolecules |
|---|---|
| Cytokines | IFNγ, IL1β, IL1Ra, IL2, IL4, IL5, IL6, IL7, IL8, IL9, IL10, IL12, IL13, IL15, IL17, TNF-α |
| Chemokines | CCL5, CCL11, IP-10, MCP-1, MIP-1α, MIP-1β |
| Growth factors | FGF-β, G-CSF, GM-CSF, PDGF-BB |
| Characteristic | Controls (n = 12) | PD (n = 16) | p Value |
|---|---|---|---|
| Age (years), mean ± SD | 75.5 ± 4.9 | 74.5 ± 8.4 | 0.6272 |
| Gender (female), n (%) | 5 (42) | 9 (38) | 0.4451 |
| BMI (kg/m2), mean ± SD | 29.2 ± 3.8 | 24.2 ± 3.0 | 0.010 |
| Number of diseases *, mean ± SD | 2.8 ± 2.1 | 3.2 ± 1.6 | 0.4621 |
| Number of medications #, mean ± SD | 2.9 ± 2.0 | 3.4 ± 1.5 | 0.3729 |
| MMSE score, mean ± SD | 27.6 ± 2.4 | 27.4 ± 2.4 | 0.8171 |
| Serum albumin (g/L), mean ± SD | 41.6 ± 7.1 | 40.3 ± 3.9 | 0.5161 |
| Total serum protein (g/L), mean ± SD | 71.8 ± 4.6 | 72.9 ± 4.8 | 0.6541 |
| Disease duration (months), mean ± SD | --- | 102.7 ± 69.1 | |
| LEDD (mg), mean ± SD | --- | 587.6 ± 223.9 |
| Controls (n = 12) | PD (n = 16) | |
|---|---|---|
| CD9 (a.u.) | 1133.4 (2710.4) | 82.3 (53.2) |
| NDUFS3 (a.u.) | 316.6 (881.8) | 96.8 ± 128.0 |
| CRP (mg/L) | 0.5 (0.7) | 1.5 (2.2) |
| FGF21 (pg/mL) | 325.3 (392.0) | 265.5 (151.8) |
| IL9 (pg/mL) | 115.0 (27.1) | 101.8 (3.6) |
| MIP-1β (pg/mL) | 158.6 (97.5) | 184.6 (23.5) |
| TNF-α (pg/mL) | 31.2 (27.4) | 42.2 (10.2) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Picca, A.; Guerra, F.; Calvani, R.; Marini, F.; Biancolillo, A.; Landi, G.; Beli, R.; Landi, F.; Bernabei, R.; Bentivoglio, A.R.; et al. Mitochondrial Signatures in Circulating Extracellular Vesicles of Older Adults with Parkinson’s Disease: Results from the EXosomes in PArkiNson’s Disease (EXPAND) Study. J. Clin. Med. 2020, 9, 504. https://doi.org/10.3390/jcm9020504
Picca A, Guerra F, Calvani R, Marini F, Biancolillo A, Landi G, Beli R, Landi F, Bernabei R, Bentivoglio AR, et al. Mitochondrial Signatures in Circulating Extracellular Vesicles of Older Adults with Parkinson’s Disease: Results from the EXosomes in PArkiNson’s Disease (EXPAND) Study. Journal of Clinical Medicine. 2020; 9(2):504. https://doi.org/10.3390/jcm9020504
Chicago/Turabian StylePicca, Anna, Flora Guerra, Riccardo Calvani, Federico Marini, Alessandra Biancolillo, Giovanni Landi, Raffaella Beli, Francesco Landi, Roberto Bernabei, Anna Rita Bentivoglio, and et al. 2020. "Mitochondrial Signatures in Circulating Extracellular Vesicles of Older Adults with Parkinson’s Disease: Results from the EXosomes in PArkiNson’s Disease (EXPAND) Study" Journal of Clinical Medicine 9, no. 2: 504. https://doi.org/10.3390/jcm9020504