
Abstract
Purpose
To determine if a regimen of restricted fluids and early vasopressor compared to usual care is feasible for initial resuscitation of hypotension due to suspected sepsis.
Methods
A prospective, randomised, open-label, clinical trial of a restricted fluid resuscitation regimen in the first 6 h among patients in the emergency department (ED) with suspected sepsis and a systolic blood pressure under 100 mmHg, after minimum 1000 ml of IV fluid. Primary outcome was total fluid administered within 6 h post randomisation.
Results
There were 99 participants (50 restricted volume and 49 usual care) in the intention-to-treat analysis. Median volume from presentation to 6 h in the restricted volume group was 2387 ml [first to third quartile (Q1–Q3) 1750–2750 ml]; 30 ml/kg (Q1–Q3 32–39 ml/kg) vs. 3000 ml (Q1–Q3 2250–3900 ml); 43 ml/kg (Q1–Q3 35–50 ml/kg) in the usual care group (p < 0.001). Median duration of vasopressor support was 21 h (Q1–Q3 9–42 h) vs. 33 h (Q1–Q3 15–50 h), (p = 0.13) in the restricted volume and usual care groups, respectively. At 90-days, 4 of 48 (8%) in the restricted volume group and 3 of 47 (6%) in the usual care group had died. Protocol deviations occurred in 6/50 (12%) in restricted group and 11/49 (22%) in the usual care group, and serious adverse events in four cases (8%) in each group.
Conclusions
A regimen of restricted fluids and early vasopressor in ED patients with suspected sepsis and hypotension appears feasible. Illness severity was moderate and mortality rates low. A future trial is necessary with recruitment of high-risk patients to determine effects on clinical outcomes in this setting.
Similar content being viewed by others


Introduction
Sepsis, defined as organ dysfunction due to infection [1], is commonly associated with hypotension due to a variable combination of peripheral vasodilatation, myocardial depression and fluid extravasation [2]. If severe, this can progress to septic shock, characterised by tissue hypoperfusion and harmful cellular and metabolic consequences. International expert consensus guidelines from the Surviving Sepsis Campaign (SSC) recommend resuscitation with at least 30 ml/kg of intravenous (IV) isotonic crystalloid over the first 3 h in patients with septic shock [3].
Despite these recommendations, there is emerging evidence of harm associated with a positive fluid balance among critically ill patients [4,5,6]. A recently published experimental study in an ovine septic shock model found IV fluid resuscitation led to a paradoxical increase in subsequent vasopressor requirement, and was associated with increases in biomarkers of cardiac stress and endothelial glycocalyx shedding [7]. Seminal clinical trials conducted in Africa found a higher mortality associated with larger volumes of IV fluid in septic shock resuscitation of adults and children [8, 9]. Translating these results to other settings is problematic, and there is currently no high level evidence to guide initial volume resuscitation in sepsis in high-income countries with ready availability of intensive care.
An alternative approach to restoring adequate blood pressure is the earlier introduction of a vasopressor infusion [10, 11]. Typically, vasopressors are commenced after between 2 and 3 l of IV fluid has been administered [12]; however earlier use may be considered ‘fluid-sparing’ [13]. Historically the requirement for a central venous catheter (CVC) posed a potential barrier to vasopressor use, but peripheral administration, at least initially, is increasingly accepted [14]. Whether a strategy of delivering a smaller volume of fluid with earlier introduction of vasopressors in adults with suspected sepsis requiring resuscitation in high-income countries is feasible remains uncertain [15, 16].
The primary aim of this pilot trial was to investigate whether, among patients assessed in the emergency department (ED) as having sepsis with hypotension, a restricted IV fluid volume and early vasopressor resuscitation protocol can achieve a clinically meaningful reduction in fluid volume compared to guideline-recommended usual care [17]. The secondary aim was to use its findings to inform the design of a future randomised clinical trial with adequate power to determine the effect of a fluid-restricted resuscitation approach on patient-centred outcomes. There is a growing rationale for such a trial [18].
Methods
The restricted fluid resuscitation in sepsis associated hypotension (REFRESH) trial was registered with the Australian and New Zealand Clinical Trials Registry (ACTRN12616000006448, 12 January 2016), and the full protocol has been previously published [19].
Design and setting
REFRESH was an investigator-initiated, multicentre, prospective, randomised open-label clinical trial with blinded outcome adjudication. Participants were recruited in the ED of eight Australian hospitals (seven teaching, one urban general) between October 2016 and March 2018.
Participants
Participants were adult patients presenting to the ED with suspected infection requiring IV antibiotic therapy who, in addition, had hypotension—defined as a systolic blood pressure (SBP) < 100 mmHg, despite the IV administration of at least 1000 ml of isotonic crystalloid fluid over a period of not more than 1 h. This SBP cut off was revised from < 90 mmHg after enrolling six cases due to a slow rate of recruitment. The full inclusion and exclusion criteria are detailed in the Supplementary Appendix.
Ethics approval and consent
Human Research Ethics Committee approval was obtained for all sites, and consent was obtained from participants or next of kin. Further details of the consent process is outlined in the Supplementary Appendix.
Randomisation
Participants were randomly assigned in a 1:1 ratio to one of the two groups, stratified by site. Randomisation was performed in real time using permuted blocks via a dedicated Web based service.
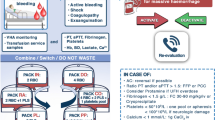
Trial interventions (Fig. 1)
The usual care treatment regimen was designed to reflect the current SSC recommendation of at least 30 ml/kg in the first 3 h. A 1000 ml bolus of isotonic crystalloid was administered (in addition to fluids given before randomisation), with further boluses of 500 ml administered hourly as required at the discretion of the treating clinician based upon routine assessment of perfusion parameters. A vasopressor infusion was commenced if the blood pressure remained inadequate despite volume loading, titrated to a target mean arterial pressure (MAP) of 65–70 mmHg. Maintenance fluids could be prescribed as required.

Summary of trial interventions. MAP mean arterial blood pressure, OP output, CVP central venous pressure
In the restricted fluid group, a vasopressor infusion was commenced if required to maintain a MAP of 65–70 mmHg. Further IV fluid boluses of 250 ml of isotonic crystalloid could be administered each hour if required at clinician discretion based upon routine assessment of perfusion parameters. Up to 1000 ml of additional fluid boluses were permitted in the restricted arm as a safety measure (e.g., clinically judged severe tachycardia, oliguria, escalating vasopressor requirement, etc.). Maintenance fluids of not more than 2 ml/kg/h could be prescribed if required.
The protocol ran for 6 h post randomisation. Beyond this time, further fluid management was determined by the treating team.
Standard treatment common to both arms
Aside from the fluid volumes, all management was as recommended in the SSC guidelines [3], including decisions regarding source control procedures. Participants received broad spectrum antibiotics directed towards the suspected source within 1 h of randomisation. Use and timing of invasive monitoring, adjunctive haemodynamic support (e.g., vasopressin, dobutamine) ventilation support, blood products and renal replacement therapy were all at the discretion of the treating team. A recommendation was made for use of balanced crystalloid solutions and use of synthetic colloid solutions was discouraged.
Outcomes
We report a range of outcomes including feasibility, process of care, and clinical measures. An embedded mechanistic study examining the effects on biomarkers of inflammation, endothelial activation and glycocalyx degradation will be reported separately [19].
Feasibility and process of care
The primary feasibility outcome was the cumulative total IV fluid volume administered at 6 h post randomisation. Secondary outcomes were total fluids administered up to 24 h, and the rates of use, time of commencement, duration and dosing, and route of administration of vasopressor drugs. We also recorded the proportion of eligible participants enrolled, rates of recruitment at each site, randomisation errors, protocol deviations and adverse events.
Clinical
Clinical outcomes were requirement for organ support [ventilation, vasopressor/inotropes, renal-replacement therapy (RRT)], sequential organ failure assessment (SOFA) score, duration of vasopressor support, peak vasopressor dose, acute kidney injury network (AKIN) score, alive organ failure ‘free days’ to day 28, intensive care unit (ICU) and hospital length of stay, alive hospital ‘free days’ to day 90, and 90-day all-cause mortality. A panel of investigators blinded to the group allocation and the fluid administration data adjudicated the clinical outcomes. As a pilot trial, REFRESH was not designed to have power to detect differences in clinical outcomes. Therefore, we report only descriptive statistics for these.
Sample size calculation
We assumed a volume in the usual care arm of 4200 ± 2650 ml within the first 6 h based upon the control group in the ARISE trial [20]. A sample size of 100 was determined to have 90% power to detect a clinically meaningful and achievable reduction of at least 30% in the restricted fluid group (to 2940 ml) compared to usual care, with two sided α = 0.05.
Data management and statistical analysis
Data were collected on paper case-report forms by research nurses or investigators and subsequently entered into a secure REDCap database [21], hosted at the University of Western Australia. Baseline data are reported as proportions for categorical variables, and as mean ± standard deviation (SD) for normally distributed, or as median with quartiles (Q1, Q3) for not normally distributed continuous variables. Primary outcomes were compared using Chi-square/Fisher’s exact test, and by Wilcoxon rank-sum test, as appropriate. We performed a post hoc sensitivity analysis excluding patients who did not meet sepsis criteria (SOFA < 2 at admission). All analyses were by intention-to-treat, and performed using Stata V14 (College Station, TX, USA).
Study management and data monitoring
The Centre for Clinical Research in Emergency Medicine at the University of Western Australia coordinated the study and a steering committee oversaw the day-to-day running of the trial. An independent Data Safety Monitoring Committee (DSMC) reviewed all adverse events (including all deaths) and evaluated the data after 50% of recruitment was complete. No formal interim analysis was undertaken.
Results
Participants
We randomised 104 patients who met all inclusion criteria. Three were ineligible (see Supplementary Appendix), one was erroneously randomised a second time, and one withdrew consent. Thus 99 participants (50 restricted volume, 49 usual care) were included in the primary analysis. The SOFA score was ≥ 2 points on admission in 93 cases; 26 cases had a SBP < 90 mmHg and lactate ≥ 2 mmol/L on admission, with 22 of these requiring vasopressors to maintain a target MAP. The flow of participants through the trial is shown in Fig. 2. Their characteristics at baseline are shown in Table 1.

Consort diagram of flow of participants through the trial
Fluid and vasopressor management
The fluid and vasopressor management over the 24-h period from arrival in the ED is shown in Table 2. Median volumes administered from ED arrival to 6 h post randomisation were 2387 ml (30 ml/kg) in the restricted volume arm, and 3000 ml (43 ml/kg) in the usual care arm (p < 0.001). At 24 h respective median cumulative volumes were 3543 ml (40 ml/kg) and 4250 ml (61 ml/kg), p = 0.005. Fluid volumes administered between 6 and 24 h did not differ significantly between the groups. Maintenance fluids were prescribed during the intervention period in 33/50 patients in the restricted volume group and in 24/49 in the usual care group. The predominant fluids used were balanced isotonic crystalloids and normal saline. Further detail of the fluid types is shown in Table S1 in the Supplementary Appendix.
There was a significant reduction in the median time from randomisation to commencement of vasopressors in the restricted fluid group (34 min vs. 150 min (p = 0.001). A higher proportion of patients in the restricted volume group had a vasopressor commenced in ED, but at 24-h there was no significant difference in the proportion receiving vasopressor support. The median duration of vasopressor infusion was 21 h in the restricted volume group vs. 33 h in the usual care group (p = 0.13); median peak vasopressor dose was 0.11 mcg/kg/min in the restricted volume group vs. 0.18 mcg/kg/min in the usual care group (p = 0.14).
Other feasibility outcomes
Recruitment rates
Four of eight sites with dedicated research staff in ED maintained screening logs during rostered staff hours. The number of potentially eligible patients presenting outside these hours and at the other sites is unknown. There were substantial differences in the total number recruited at each site (Figs. S1 and S2 and Table S2 in the Supplementary Appendix).
Protocol compliance
Six participants in the usual care group did not receive the minimum prescribed volume of 1000 ml within the 6-h intervention period; one participant in the restricted volume group received in excess of 3000 ml during the intervention period; five participants (three usual care, two restricted) were transferred to the operating theatre within 6 h requiring discontinuation of the protocol. Further details on protocol deviations are provided in Table S3 in the Supplementary Appendix.
Adverse events
There were four adverse events recorded in each group (Table S4 in the Supplementary Appendix). The DSMC reviewed all deaths, and all were determined to have been due to underlying disease, with participation in the trial not a contributing factor.
Clinical outcomes
Clinical outcomes for the trial participants are shown in Table 3. Rates of ICU admission were 33/50 (66%) and 29/49 (59%); rates of ventilation were 10/50 (20%) and 9/49 (18%); rates of RRT were 4/50 (8%) and 4/49 (8%) in the restricted and usual care groups, respectively. SOFA scores are shown in Figs. S3 and S4 in the Supplementary Appendix. At 90-days 4/48 (8%) participants in the restricted volume group and 3/47 (6%) in the usual care group had died. Further details on those who died are provided in Table S5 in the Supplementary Appendix.
Sensitivity analysis of cases meeting sepsis criteria
The results of a sensitivity analysis of participants with sepsis, i.e., admission SOFA score ≥ 2 (N = 93) are shown in Tables S6 and S7 in the Supplementary Appendix. The findings were consistent with the primary analysis.
Discussion
Key findings
We demonstrated that a restricted volume/early vasopressor approach over the first 6 h of resuscitation in patients presenting to the ED with suspected sepsis and hypotension resulted in a 30% relative reduction in total fluid administered up to 24 h and was not associated with any signal of harm. Despite a higher proportion of patients in the restricted volume group receiving vasopressors early in the ED, this did not result in a longer overall duration of vasopressor use in this group.
Relationship to other studies
There is accumulating evidence of harm associated with a positive fluid balance in ICU patients with sepsis [22,23,24,25]. A multicentre, pilot clinical trial of a fluid sparing regimen among ICU patients with septic shock in Scandinavia found lower rates of acute kidney injury [26]; however the median volume of fluid administered prior to randomisation was over 4 l. To our knowledge ours is the first clinical trial outside Africa to compare an IV fluid volume restricted and early vasopressor regimen to the SSC recommended approach [3] for initial resuscitation of adults presenting to the ED with suspected sepsis and hypotension. A phase III clinical trial addressing this question has recently commenced in the USA [18].
Meaning of the study
A recent large, randomised trial investigated the role of a perioperative fluid-sparing regimen for patients undergoing major elective abdominal surgery [27]. Contrary to expectations based upon previous smaller studies, this approach was associated with a higher incidence of acute kidney injury. While not translatable to sepsis resuscitation, this cautions against the premature adoption of fluid-sparing management strategies based upon a limited evidence base, and emphasises the importance of large scale, rigorously conducted clinical trials to inform practice. Testing the question of IV fluid volume in sepsis resuscitation has been identified as a top research priority in critical care [28]. We have demonstrated that such a trial intervention is feasible.
Strengths and limitations
The strengths of this pilot trial are multicentre design, pragmatic enrolment criteria, a clinically informed and acceptable protocol resulting in meaningful separation, the collection of patient centred outcome and safety data and high rates of completed follow up.
There are several limitations. The overall mortality rate of 7% was lower than expected. This may be related to the SBP < 100 mmHg inclusion criterion. This is consistent with the blood pressure threshold in the Sepsis 3 task force ‘qSOFA’ score [1], however only a minority of participants met criteria for septic shock. Further explanations may be the exclusion of patients likely to require urgent surgery, such as intra-abdominal sepsis, which typically has a higher mortality. There were more protocol deviations relating to fluid volume in the usual care group than in the restricted fluid group. This may reflect a bias among clinicians in favour of restricting IV fluid in this un-blinded trial. Finally, there are challenges inherent in recruiting the most sick and unstable patients within a limited time-window in the ED. A recently published pilot trial of restricted fluid volume resuscitation of paediatric sepsis patients in the UK also found a lower than expected severity of illness [29].
Despite achieving a 30% relative reduction in fluid volume, the absolute difference between the groups was less than 1 l. Whether this difference is clinically meaningful is uncertain, although in a small Zambian clinical trial, a difference in median fluid volumes at 24 h of 4 l vs. 3 l was associated with a significant reduction in mortality in the lower volume group [9]. The median cumulative volume at 6 h in the usual care arm of our trial was 3000 ml, less than the volume of over 4000 ml administered in three international early goal directed therapy trials conducted in the past decade [12]. Whether this reflects the lower illness severity, or an incremental change in routine practice is unknown.
Unanswered questions and implications for a future trial
While the trial achieved the feasibility objectives, there are important implications for designing a future clinical trial with sufficient outcome event rates to detect differences in these rates. The low mortality rate could be addressed by more stringent inclusion criteria targeted to sicker patients (e.g., established septic shock). Requiring a larger fluid volume prior to being eligible would select for more severe illness, but could threaten separation between the experimental groups. Including patients with intra-abdominal sepsis and developing a peri-operative protocol would increase the potential recruitment pool, and allow higher risk patients to be enrolled.
It is possible that greater separation in fluid volumes could be achieved by mandating a larger ‘minimum’ volume in the control arm, while ensuring flexibility for clinicians to manage patients in accordance with usual care. Conversely, maintenance fluid should be prohibited in the restricted fluid would since this accounts for a substantial proportion of total IV fluid volume in critically ill patients [30]. Any future trial with mortality and other clinical endpoints would require a sample size informed by a thorough understanding of contemporary ED practice relating to fluid volume and timing of vasopressors, and the epidemiology and mortality rates for the cohort of interest.
Conclusions
For patients presenting to the ED with suspected sepsis and hypotension, a fluid-restricted and early vasopressor regimen resulted in a reduction in total fluid volume administered in the first 24 h. There was no increase in overall duration of vasopressor use and no evidence of harm. A large clinical trial to investigate the effects of such an approach to resuscitation on patient centred outcomes appears feasible. Modifications to the present protocol would be required to maximise meaningful separation between the study groups, and recruitment of high-risk patients to deliver sufficient statistical power to determine effects on mortality in this setting.
References
Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, Hotchkiss RS, Levy MM, Marshall JC, Martin GS, Opal SM, Rubenfeld GD, van der Poll T, Vincent JL, Angus DC (2016) The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA 315:801–810
Angus DC, van der Poll T (2013) Severe sepsis and septic shock. N Engl J Med 369:840–851
Rhodes A, Evans LE, Alhazzani W, Levy MM, Antonelli M, Ferrer R, Kumar A, Sevransky JE, Sprung CL, Nunnally ME, Rochwerg B, Rubenfeld GD, Angus DC, Annane D, Beale RJ, Bellinghan GJ, Bernard GR, Chiche JD, Coopersmith C, De Backer DP, French CJ, Fujishima S, Gerlach H, Hidalgo JL, Hollenberg SM, Jones AE, Karnad DR, Kleinpell RM, Koh Y, Lisboa TC, Machado FR, Marini JJ, Marshall JC, Mazuski JE, McIntyre LA, McLean AS, Mehta S, Moreno RP, Myburgh J, Navalesi P, Nishida O, Osborn TM, Perner A, Plunkett CM, Ranieri M, Schorr CA, Seckel MA, Seymour CW, Shieh L, Shukri KA, Simpson SQ, Singer M, Thompson BT, Townsend SR, Van der Poll T, Vincent JL, Wiersinga WJ, Zimmerman JL, Dellinger RP (2017) Surviving sepsis campaign: international guidelines for management of sepsis and septic shock: 2016. Intensive Care Med 43:304–377
Silversides JA, Major E, Ferguson AJ, Mann EE, McAuley DF, Marshall JC, Blackwood B, Fan E (2017) Conservative fluid management or deresuscitation for patients with sepsis or acute respiratory distress syndrome following the resuscitation phase of critical illness: a systematic review and meta-analysis. Intensive Care Med 43:155–170
Perner A, Cecconi M, Cronhjort M, Darmon M, Jakob SM, Pettila V, van der Horst ICC (2018) Expert statement for the management of hypovolemia in sepsis. Intensive Care Med 44:791–798
National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network, Wiedemann HP, Wheeler AP, Bernard GR, Thompson BT, Hayden D, deBoisblanc B, Connors AF Jr., Hite RD, Harabin AL (2006) Comparison of two fluid-management strategies in acute lung injury. N Engl J Med 354:2564–2575
Byrne L, Obonyo NG, Diab SD, Dunster KR, Passmore MR, Boon AC, See Hoe L, Pedersen S, Hashairi Fauzi M, Pretti Pimenta L, Van Haren F, Anstey CM, Cullen L, Tung JP, Shekar K, Maitland K, Fraser JF (2018) Unintended consequences; fluid resuscitation worsens shock in an ovine model of endotoxemia. Am J Respir Crit Care Med. https://doi.org/10.1164/rccm.201801-0064oc
Maitland K, Kiguli S, Opoka RO, Engoru C, Olupot-Olupot P, Akech SO, Nyeko R, Mtove G, Reyburn H, Lang T, Brent B, Evans JA, Tibenderana JK, Crawley J, Russell EC, Levin M, Babiker AG, Gibb DM, on behalf of the FEAST trial group (2011) Mortality after fluid olus in Africal children with severe infection. N Engl J Med 364:2483–2495
Andrews B, Semler MW, Muchemwa L, Kelly P, Lakhi S, Heimburger DC, Mabula C, Bwalya M, Bernard GR (2017) Effect of an early resuscitation protocol on in-hospital mortality among adults with sepsis and hypotension. JAMA 318:1233–1240
Waechter J, Kumar A, Lapinsky SE, Marshall J, Dodek P, Arabi Y, Parrillo JE, Dellinger RP, Garland A, Cooperative Antimicrobial Therapy of Septic Shock Database Research Group (2014) Interaction between fluids and vasoactive agents on mortality in septic shock: a multicenter, observational study. Crit Care Med 42:2158–2168
Bai X, Yu W, Ji W, Lin Z, Tan S, Duan K, Dong Y, Xu L, Li N (2014) Early versus delayed administration of norepinephrine in patients with septic shock. Crit Care 18:532
Rowan KM, Angus DC, Bailey M, Barnato AE, Bellomo R, Canter RR, Coats TJ, Delaney A, Gimbel E, Grieve RD, Harrison DA, Higgins AM, Howe B, Huang DT, Kellum JA, Mouncey PR, Music E, Peake SL, Pike F, Reade MC, Sadique MZ, Singer M, Yealy DM, the PRISM Investigators (2017) Early, goal-directed therapy for septic shock: a patient-level meta-analysis. N Engl J Med 376:2223–2234
Hamzaoui O, Georger JF, Monnet X, Ksouri H, Maizel J, Richard C, Teboul JL (2010) Early administration of norepinephrine increases cardiac preload and cardiac output in septic patients with life-threatening hypotension. Crit Care 14:R142
Hallengren M, Astrand P, Eksborg S, Barle H, Frostell C (2017) Septic shock and the use of norepinephrine in an intermediate care unit: mortality and adverse events. PLoS One 12:e0183073
Byrne L, Van Haren F (2017) Fluid resuscitation in human sepsis: time to rewrite history? Ann Intensive Care 7:4
Finfer S, Myburgh J, Bellomo R (2018) Intravenous fluid therapy in critically ill adults. Nat Rev Nephrol 14:541–557
Macdonald SPJ, Keijzers G, Taylor DM, Kinnear FB, Arendts G, Fatovich DM, Bellomo R, McCutcheon D, Fraser JF, Burrows S, Ascencio-Lane JC, Litton E, Harley A, Anstey M, Mukherjee A (2018) Restricted fluid resuscitation in sepsis associated hypotension (REFRESH): a propsective, multicentre, clinical feasibility trial. Intensive Care Med Exp 6:21 (Abstract presented at Sepsis 2018, Bangkok Thailand, 1 October 2018)
Self WH, Semler MW, Bellomo R, Brown SM, deBoisblanc BP, Exline MC, Ginde AA, Grissom CK, Janz DR, Jones AE, Liu KD, Macdonald SPJ, Miller CD, Park PK, Reineck LA, Rice TW, Steingrub JS, Talmor D, Yealy DM, Douglas IS, Shapiro NI, CLOVERS Protocol Committee and NHLBI Prevention and Early Treatment of Acute Lung Injury (PETAL) Network Investigators (2018) Liberal versus restrictive intravenous fluid therapy for early septic shock: rationale for a randomized trial. Ann Emerg Med 72:457–466
Macdonald SPJ, Taylor DM, Keijzers G, Arendts G, Fatovich DM, Kinnear FB, Brown SGA, Bellomo R, Burrows S, Fraser JF, Litton E, Ascencio-Lane JC, Anstey M, McCutcheon D, Smart L, Vlad I, Winearls J, Wibrow B (2017) Restricted fluid resuscitation in sepsis-associated hypotension (REFRESH): study protocol for a pilot randomised controlled trial. Trials 18:399
Peake SL, Delaney A, Bailey M, Bellomo R, Cameron PA, Cooper DJ, Higgins AM, Holdgate A, Howe BD, Webb SA, Williams P, the ARISE Investigators (2014) Goal-directed resuscitation for patients with early septic shock. N Engl J Med 371:1496–1506
Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG (2009) Research electronic data capture (REDCap)—A metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform 42:377–381
Kelm DJ, Perrin JT, Cartin-Ceba R, Gajic O, Schenck L, Kennedy CC (2015) Fluid overload in patients with severe sepsis and septic shock treated with early goal-directed therapy is associated with increased acute need for fluid-related medical interventions and hospital death. Shock 43:68–73
Acheampong A, Vincent JL (2015) A positive fluid balance is an independent prognostic factor in patients with sepsis. Crit Care 19:251
Sirvent JM, Ferri C, Baro A, Murcia C, Lorencio C (2015) Fluid balance in sepsis and septic shock as a determining factor of mortality. Am J Emerg Med 33:186–189
Marik PE, Linde-Zwirble WT, Bittner EA, Sahatjian J, Hansell D (2017) Fluid administration in severe sepsis and septic shock, patterns and outcomes: an analysis of a large national database. Intensive Care Med 43:625–632
Hjortrup PB, Haase N, Bundgaard H, Thomsen SL, Winding R, Pettila V, Aaen A, Lodahl D, Berthelsen RE, Christensen H, Madsen MB, Winkel P, Wetterslev J, Perner A, CLASSIC Trial Group, Scandinavian Critical Care Trials Group (2016) Restricting volumes of resuscitation fluid in adults with septic shock after initial management: the CLASSIC randomised, parallel-group, multicentre feasibility trial. Intensive Care Med 42:1695–1705
Myles PS, Bellomo R, Corcoran T, Forbes A, Peyton P, Story D, Christophi C, Leslie K, McGuinness S, Parke R, Serpell J, Chan MTV, Painter T, McCluskey S, Minto G, Wallace S, Australian and New Zealand College of Anaesthetists Clinical Trials Network and the Australian and New Zealand Intensive Care Society Clinical Trials Group (2018) Restrictive versus liberal fluid therapy for major abdominal surgery. N Engl J Med 378:2263–2274
Perner A, Gordon AC, Angus DC, Lamontagne F, Machado F, Russell JA, Timsit JF, Marshall JC, Myburgh J, Shankar-Hari M, Singer M (2017) The intensive care medicine research agenda on septic shock. Intensive Care Med 43:1294–1305
Inwald DP, Canter R, Woolfall K, Mouncey P, Zenasni Z, O’Hara C, Carter A, Jones N, Lyttle MD, Nadel S, Peters MJ, Harrison DA, Rowan KM, PERUKI (Paediatric Emergency Research in the UK and Ireland) and PICS SG (Paediatric Intensive Care Society Study Group) (2018) Restricted fluid bolus volume in early septic shock: results of the fluids in shock pilot trial. Arch Dis Child. https://doi.org/10.1136/archdischild-2018-314924 (Epub ahead of print)
Van Regenmortel N, Verbrugghe W, Roelant E, Van den Wyngaert T, Jorens PG (2018) Maintenance fluid therapy and fluid creep impose more significant fluid, sodium, and chloride burdens than resuscitation fluids in critically ill patients: a retrospective study in a tertiary mixed ICU population. Intensive Care Med 44:409–417
Acknowledgements
REFRESH trial investigators and participating sites: Armadale Health Service, WA (David McCutcheon, Ashes Mukherjee, Anton Leonard, Jonathan Burcham); Austin Health, VIC (David Taylor, Rinaldo Bellomo); Fiona Stanley Hospital, WA (Glenn Arendts, Edward Litton); Gold Coast University Hospital, QLD (Gerben Keijzers, Amanda Harley, James Winearls); Royal Hobart Hospital (Juan Carlos Ascencio-Lane, Simon Brown, David Cooper); Royal Perth Hospital/Centre for Clinical Research in Emergency Medicine, WA (Stephen Macdonald [steering committee chair], Daniel Fatovich, Lisa Smart); Sir Charles Gairdner Hospital, WA (Ioana Vlad, Bradley Wibrow, Matthew Anstey); The Prince Charles Hospital, QLD (Frances Kinnear, John Fraser). Trial Management and Coordination: Ellen Macdonald, Sophie Damianopoulos. Data Safety Monitoring Committee: Anthony Brown, Robert Boots, Michael Phillips. Trial endorsed by the Australasian College for Emergency Medicine Clinical Trials Group.
Funding
The REFRESH trial was an investigator-initiated study funded in part by a grant from the Emergency Medicine Foundation, Queensland, Australia EMSS-229R24-2015, and supported by the participating institutions.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflicts of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Macdonald, S.P.J., Keijzers, G., Taylor, D.M. et al. Restricted fluid resuscitation in suspected sepsis associated hypotension (REFRESH): a pilot randomised controlled trial. Intensive Care Med 44, 2070–2078 (2018). https://doi.org/10.1007/s00134-018-5433-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00134-018-5433-0